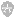微生物的分类单元和命名
2009-12-16 22:10:25 来源:本站原创 评论:0 点击:
分类是人类认识微生物,进而利用和改造微生物的一种手段,微生物工作者只有在掌握了分类学知识的基础上,才能对纷繁的微生物类群有一清晰的轮廊,了解其亲缘关系与演化关系,为人类开发利用微生物资源提供依据。
微生物分类学 (microbial taxonomy) 是一门按微生物的亲缘关系把它们安排成条理清楚的各种分类单元或分类群 (taxon) 的科学,它的具体任务有三,即分类 (classification) 、命名 (nomenclature) 和鉴定 (identification) 。分类指的是根据相似性或亲缘关系,将一个有机体放在一个单元中。命名是按照国际命名法规给有机体一个科学名称。鉴定则是确定一个新的分离物是否归属于已经命名的分类单元的过程。因此,概括来说,微生物分类学是对各个微生物进行鉴定,按分类学准则排列成分类系统,并对已确定的分类单元进行科学命名的科学。
微生物的主要分类单位,依次为界 (kingdom) 、门( phylum 或 division )、纲 (class) 、目 (order) 、科 (fami1y) 、属 (genus) 、种 (species) 。其中种是最基本的分类单位。具有完全或极多相同特点的有机体构成同种。性质相似、相互有关的各种组成属。相近似的届合并为科。近似的科合并为目。近似的目归纳为纲。综合各纲成为门。由此构成一个完整的分类系统。以下以柠檬浮霉状菌为例加以说明。
 |
另外,每个分类单位都有亚级,即在两个主要分类单位之间,可添加“亚门”、“亚纲”、“亚目、”“亚科”等次要分类单位。在种以下还可以分为亚种、变种、型、菌株等。
属是科与种之间的分类单元,通常包含具有某些共同特征和关系密切的种。 Goodfellow 和 O"Donnell(1993) 提出 DNA 的 G+C mol% 差异≤ 10 % ~12 %及 16S rDNA 的序列同源性≥ 95 %的种可归为同一属。
种 (species) 关于微生物“种”的概念,各个分类学家的看法不一,例如伯杰氏 (Bergey) 给种的定义是:“凡是与典型培养菌密切相同的其他培养菌统一起来,区分成为细菌的一个种。”因此,它是以某个“标准菌株”为代表的十分类似的菌株的总体。种是以群体形式存在的。种有着不同的定义,在微生物学中较常见有生物学种( biological species , BS ),进化种 (evolutionary species , ES) 和系统发育种( phylogenetic species , PS )等不同的物种概念。
1986 年斯坦尔 (Stanier) 给种下了定义:“一个种是由一群具有高度表型相似性的个体组成,并与其他具有相似特征的类群存在明显的差异。”但这个定义仍无量化标准。 1987 年,国际细菌分类委员会颁布, DNA 同源性≥ 70 %,而且其⊿ T m ≤ 5 ℃的菌群为一个种,并且其表型特征应与这个定义相一致。 1994 年 Embley 和 Stackebrandt 认为当 16S rDNA 的序列同源性≥ 97 %时可认为是一个种。
亚种 (subspecies) 在种内,有些菌株如果在遗传特性上关系密切,而且在表型上存在较小的某些差异,一个种可分为两个或两个以上小的分类单位,称为亚种。它们是细菌分类中具有正式分类地位的最低等级。根据⊿ T m 值在 DNA 杂交中的频率分布,有些证据表明,亚种的概念在系统发育上是有效的,而且能与亚种以下的变种概念相区别。后者仅是依据所选择的“实用”属性而决定,并不被 DNA 组成所证明。
亚种以下的分类等级 通常表示能用某些特殊的特征加以区别的菌株类群。例如,在细菌分类中,以生物变型 (biovar) 表示特殊的生化或生理特征,血清变型 (serovar 结构的不同,致病变型 (pathovar) 表示某些寄主的专一致病性,噬菌变型 (phagovar) 表示对噬菌体的特异性反应,形态变型 (morphovar) 表示特殊的形态特征。
菌株或品系 (strain) 这是微生物学中常碰到的一个名词,它主要是指同种微生物不同来源的纯培养物。从自然界分离纯化所得到的纯培养的后代,经过鉴定属于某个种,但由于来自不同的地区、土壤和其他生活环境,它们总会出现一些细微的差异。这些单个分离物的纯培养的后代称为菌株。菌株常以数目、字母、人名或地名表示。那些得到分离纯化而未经鉴定的纯培养的后代则称为分离物。
微生物学中还常常用到“群”这个词,这只是为了科研或鉴定工作方便,首先按其形态或结合少量的生理生化、生态学特征,将近似的种和介于种间的菌株归纳为若干个类群。如为了筛选抗生素工作的方便,中国科学院微生物研究所根据形态和培养特征,把放线菌中的链霉菌属归纳为 12 个类群。
微生物分类各级单元所用的后缀如表。
表 14 - 1 微生物分类各级单元拉丁学名后缀
 |
二、微生物的命名
微生物的命名和其他生物一样,都按国际命名法命名,即采用林奈氏 (Linnaeus) 所创立的“双名法”。每一种微生物的学名都依属与种而命名,由两个拉丁字或希腊字或者拉丁化了的其他文字组成。属名在前,规定用拉丁字名词表示,字首字母要大写,由微生物的构造、形状,或由著名的科学家名字而来,用以描述微生物的主要特征。种名在后,用拉丁字形容词表示,字首字母小写,为微生物的色素、形状、来源、病名或著名的科学家姓名等,用以描述微生物的次要特征。此外,由于自然界的生物种类太多了,大家都在命名,为了更明确,避免误解,故在正式的拉丁名称后面附着命名者的姓。例如。金黄色葡萄球菌的学名为:
Staphylococcus aureus Rosenbach 1884
属名:葡萄球菌 种名:金黄色 命名人的姓 命名年份
又如: Peptostreptococcus foetidus ( Veillon ) Smith
属名:消化链球菌 种名:恶臭 原命名者 改名者
恶臭消化链球菌是由 Veillon 首先发现和定名的,后 Smith 重新定为现名。由此可以看出,种名后括弧内的姓,是表示这个种首先由 Veillon 定的名,在括弧后再附加改定此菌学名人的姓。如果发表新种,则在学名之后加 n . sp( 即 novo species 的缩写,意为新种 ) 。有时只泛指某一属的微生物,而不是指定某一个具体的种,或没有种名,只有属名时,可在属名后加 sp. 或 spp.(species 的缩写, sp. 表示单数, spp. 表示复数 ) ,如 Micrococcus sp. ,表示微球菌属的一个种, Micrococcus spp. 表示微球菌属的一些种。变种的学名,是在种名后加变种名称,并在变种名称之前加 var 如枯草芽孢杆菌黑色变种应写成 Bacillus subtilis var. niger 。 属以上的名称必须是阴性复数形容词,与 prokaryotae 相一致。
由于现代分子生物学技术的迅速发展,正在形成一套与传统的分类鉴定方法完全不同的分类鉴定技术与方法,从基因水平上分析各微生物种之间的亲缘关系,即系统发育地位。众所周知,原核生物细胞中的 16S rDNA 和真核生物细胞中的 18S rDNA 的碱基序列都是十分保守的,不受微生物所处环境条件的变化、营养物质的丰缺的影响而有所变化,都可以看作为生物进化的时间标尺,记录着生物进化的真实痕迹。因此,分析原核生物细胞中的 16S rDNA 和真核生物细胞中的 18S rDNA 的碱基序列,比较所分析的微生物与其他微生物种之间 16S rDNA 和 18S rDNA 序列的同源性,可以真实地揭示它们亲缘关系的距离和系统发育地位。在现实研究中,除了选择 16S rDNA 和 18S rDNA 作序列分析进行系统发育比较外,还可利用间隔序列 (ITS) 、某些发育较为古老而序列又较稳定的特异性酶的基因作序列分析,进行系统发育分析。如在环境微生物研究中,对于谷胱甘肽转移酶( GST )的基因序列分析所获得的系统发育鉴定结果与用其他方法所获得的结果具有十分吻合的一致性。随着研究技术和理论的日趋成熟,现在有人提出了分子系统学 (molecular systematics) 这一理论概念。
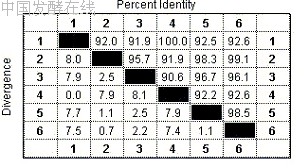
系统学( systematics )是研究生物多样性及其分类和演化关系的科学。分子系统学是检测、描述并揭示生物在分子水平上的多样性及其演化规律的科学。研究内容包括了群体遗传结构、分类学、系统发育和分子进化等领域。群体遗传结构 (population genetic structure) 是指一个种内总的遗传变异程度及其在群体间的分布模式,是一个种最基础的遗传信息。分类学 (taxonomy) 是研究物种的界定和序级确定。系统发育关系 (phylogenetic relationship) 和分子进化 (molecular evolution) 是两个密切相关的过程。在利用现代分子生物学技术在分子和基因水平上获得大量的分类单元尤其是种的遗传信息后,来推断和重建微生物类群的演化历史和演化关系,即建立系统发育树,如第一章中图 1-1 表示细菌、古菌和真核生物的无根系统发育树。根据分离菌株的 16S rDNA 或 18S rDNA 序列与相关微生物种之间的同源性,将分离获得的菌株放置于系统发育树的确当分支位置,以显示其在系统发育中的地位和与其他种间的亲缘关系。原核微生物中的细菌和古菌的系统发育树分别如图 14-1 和 14-2 所示。
 |
图 14-1 细菌域的系统发育树(引自 Madigan et al., Brock Biology of Microorganisms, Tenth edition, 2003 )
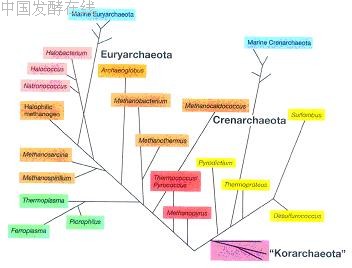 |
图 14-2 古菌域的系统发育树(引自 Madigan et al., Brock Biology of Microorganisms, Tenth edition, 2003 )
微生物的分类鉴定方法
一、微生物鉴定的依据
获得纯化的微生物分离菌株后,首先判定是原核微生物还是真核微生物,这实际上在分离过程中所使用的方法和选择性培养基已经决定了分离菌株的大类的归属,从平板菌落的特征和液体培养的性状都可加以判定。然后,如是原核微生物,便可根据表 14-3 所示的经典分类鉴定指标进行鉴定,如条件允许,可做碳源利用的 BIOLOG-GN 分析和 16S rDNA 序列分析。多项结果结合起来确定分离菌株的属和种。
表 14-3 微生物经典分类鉴定方法的指标依据
|
经 典 分 类 法 |
个体:细胞形态、大小、排列方式,染色反应,有无运动,各种特殊构造特征等 |
|
形态特征:菌落形态,在固体、半固体或液体培养基中的生长状态等 | |
|
营养要求:碳源、氮源、矿质元素、生长因子等 | |
|
生理生化特征:代谢产物种类、产量、显色反应等 | |
|
酶:产酶种类和反应特征等 | |
|
生态学特性:生长温度,对氧的需要,酸碱度要求,宿主种类,生态分布等 | |
|
血清学反应 | |
|
噬菌体的敏感性 | |
|
其他 | |
根据目前微生物分类学中使用的技术和方法,可把它们分成四个不同的水平:①细胞形态和行为水平,②细胞组分水平,③蛋白质水平,④基因组水平;
在微生物分类学发展的早期,主要的分类鉴定指标是以在细胞形态和习性为主,可称为经典的分类鉴定法。其他三种实验技术主要是 60 年代以后采用的,称为化学分类和遗传学分类法,这些方法再加上数值分类鉴定法,可称为现代的分类鉴定方法。
(一)、经典分类鉴定法
经典分类法是一百多年来进行微生物分类的传统方法。其特点是人为地选择几种形态生理生化特征进行分类,并在分类中将表型特征分为主、次。一般在科以上分类单位以形态特征、科以下分类单位以形态结合生理生化特征加以区分。最后,采用双歧法整理实验结果,排列一个个的分类单元,形成双歧检索表(图 14-4 )。
A. 能在 60 o C 以上生长
B. 细胞大,宽度 1.3~1.8mm ……………………………………… 1. 热微菌属 ( Thermomicrobium )
BB. 细胞小,宽度 0.4~0.8mm
C. 能以葡萄糖为碳源生长
D. 能在 pH4.5 生长 …………………………………………… 2. 热酸菌属 ( Acidothermus )
DD. 不能在 pH4.5 生长 ………………………………………………… 3. 栖热菌属 ( Thermus )
CC. 不能以葡萄糖为唯一碳源 ……………………… 4. 栖热嗜油菌属 ( 栖热嗜狮菌属 Thermoleophilum )
AA. 不能在 60 o C 以上生长
图 14-4 双歧法检索表例样
应用 BIOLOG-GN 仪检测分离菌株对众多碳源的利用情况判断分离菌株的分类地位,近年来也时有应用。在 BIOLOG-GN 仪上有 96 个小孔,其中 95 孔内分装有 95 种不同碳源的缓冲液, 1 孔为无碳源的缓冲液对照,各孔接入适宜菌浓度和液量的分离菌株培养物,定温培养,每日定时读取 BIOLOG-GN 仪计算机上各碳源利用情况,一般为时 1 周, BIOLOG-GN 仪可显示出该鉴定菌株的最可能归属。
(二)、数值分类法
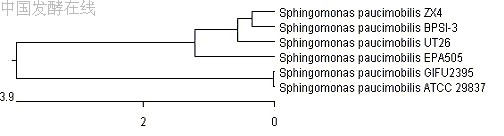
又称阿德逊氏分类法 () 。它的特点是根据较多的特征进行分类,一般为 50 ~ 60 个,多者可达 100 个以上,在分类上,每一个特性的地位都是均等重要。通常是以形态、生理生化特征,对环境的反应和忍受性以及生态特性为依据。最后,将所测菌株两两进行比较,并借用电子计算机计算出菌株间的总相似值,列出相似值矩阵 ( 图 14-5) 。为便于观察,应将矩阵重新安排,使相似度高的菌株列在一起,然后将矩阵图转换成树状谱 (dendrogram)( 图 14-6) ,再结合主观上的判断 ( 如划分类似程度大于 85 %者为同种,大于 65 %者为同属等 ) ,排列出—个个分类群。
|
图 14-5 显示 6 个细菌菌株的遗传相似矩阵图
|
图 14-6 根据相似矩阵图转换的相似关系树状谱
数值分类法的优越性在于它是以分析大量分类特征为基础,对于类群的划分比较客观和稳定;而且促进对细菌类群的全面考查和观察,为细菌的分类鉴定积累大量资料。但在使用数值分类法对细菌菌株分群归类定种或定属时,还应做有关菌株的 DNA 碱基的 G + Cmol% 和 DNA 杂交,以进一步加以确证。
( 三)、化学分类法
微生物分类中,根据微生物细胞的特征性化学组分对微生物进行分类的方法称化学分类法 (chemotaxonomy) 。在近二十多年中,采用化学和物理技术采研究细菌细胞的化学组成,已获得很有价值的分类和鉴定资料,各种化学组分在原核微生物分类中的意义见表 14-4 。
表 14-4 细菌的化学组分分析及其在分类水平上的应用
|
细胞成份 |
分析内容 |
在分类水平上的作用 |
|
细胞壁 |
肽聚糖结构 |
种和属 |
|
多糖 | ||
|
胞壁酸 | ||
|
膜 |
脂肪酸 |
种和属 |
|
极性类脂 | ||
|
霉菌酸 | ||
|
类异戊二烯苯醌 | ||
|
蛋白质 |
氨基酸序列分析 |
属和属以上单位 |
|
血清学比较 | ||
|
电泳图 | ||
|
酶谱 | ||
|
代谢产物 |
脂肪酸 |
种和属 |
|
全细胞成分分析 |
热解—气液色谱分析 |
种和亚种 |
|
热解 — 质谱分析 |
随着分子生物学的发展,细胞化学组分分析用于微生物分类日趋显示出重要性。细胞壁的氨基酸种类和数量现己被接受为细菌属的水平的重要分类学标准。在放线菌分类中,细胞壁成分和细胞特征性糖的分析作为分属的依据,已被广泛应用。脂质是区别细菌还是古菌的标准之一,细菌具有酰基脂 ( 脂键 ) ,而古菌具有醚键脂,因此醚键脂的存在可用以区分古菌。霉菌酸的分析测定己成为诺卡氏菌形放线菌分类鉴定中的常规方法之一。鞘氨醇单胞菌和鞘氨醇杆菌等细胞膜都含有鞘氨醇,因此鞘氨醇的有无可作为此类细菌的一个重要标志。此外某些细菌原生质膜中的异戊间二烯醌,细胞色素,以及红外光谱等分析对于细菌、放线菌中某些科、属、种的鉴定也都十分有价值。
(四)、遗传学分类法
分子遗传学分类法是以微生物的遗传型 ( 基因型 ) 特征为依据,判断微生物问的亲缘关系,排列出一个个的分类群。目前较常使用的方法有:
1 、 DNA 中 G+C mol% 分析
每一个微生物种的 DNA 中 GC mol% 的数值是恒定的,不会随着环境条件、培养条件等的变化而变化,而且在同一个属不同种之间, DNA 中 GCmol% 的数值不会差异太大,可以某个数值为中心成簇分布,显示同属微生物种的 GC mol% 范围。 DNA 中 GC mol% 分析主要用于区分细菌的属和种,因为细菌 DNA 中 GC 含量的变化范围一般在 25 %~ 75 %;而放线菌 DNA 中的 GC 比例范围非常窄 (37 %~ 51%) 。一般认为任何两种微生物在 GC 含量上的差别超过了 10 %,这两种微生物就肯定不是同一个种。因此可利用 G+C mol %来鉴别各种微生物种属间的亲缘关系及其远近程度。值得注意的是,亲缘关系相近的菌,其 G+C mol %含量相同或者近似,但 G+C mol %相同或近似的细菌,其亲缘关系并不一定相似,这是因为这一数据还不能反映出碱基对的排列序列,而且如放线菌的 DNA 的 GC mol% 在 37 ~ 51 之间,企图在这么小的范围内区分放线菌的几十个属显然是不现实的。要比较两种细菌的 DNA 碱基对排列序列是否相同,以及相同的程度如何,就需做核酸杂交试验。
2 、 DNA-DNA 杂交
DNA 杂交法的基本原理是用 DNA 解链的可逆性和碱基配对的专一性,将不同来源的 DNA 在体外加热解链,并在合适的条件下,使互补的碱基重新配对结合成双链 DNA ,然后根据能生成双链的情况,检测杂合百分数。如果两条单链 DNA 的碱基顺序全部相同,则它们能生成完整的双链,即杂合率为 100% 。如果两条单链 DNA 的碱基序列只有部分相同,则它们能生成的“双链”仅含有局部单链,其杂合率小于 100% 。由此;杂合率越高,表示两个 DNA 之间碱基序列的相似性越高,它们之间的亲缘关系也就越近。如两株大肠埃希氏菌的 DNA 杂合率可高达 100 %,而大肠埃希氏菌与沙门氏菌的 DNA 杂合率较低,约有 70 %。 G+Cmol %的测定和 DNA 杂交实验为细菌种和属的分类研究开辟了新的途径,解决了以表观特征为依据所无法解决的一些疑难问题,但对于许多属以上分类单元间的亲缘关系及细菌的进化问题仍不能解决。
3 、 DNA — rRNA 杂交
目前研究 RNA 碱基序列的方法有两种。一是 DNA 与 rRNA 杂交,二是 16S rRNA 寡核苷酸的序列分析。 DNA 与 rRNA 杂交的基本原理、实验方法同 DNA 杂交一样,不同的是① 是 DNA 杂交中同位素标记的部分是 DNA ,而 DNA 与 rRNA 杂交中同位素标记的部分是 rRNA 。② DNA 杂交结果用同源性百分数表示,而 DNA 与 rRNA 杂交结果用 Tm(e) 和 RNA 结合数表示。 Tm(e) 值是 DNA 与 rRNA 杂交物解链一半时所需要的温度。 RNA 结合数是 100 m gDNA 所结合的 rRNA 的 m g 数。根据这个参数可以作出 RNA 相似性图。在 rRNA 相似性图上,关系较近的菌就集中到一起。关系较远的菌在图上占据不同的位置。用 rRNA 同性试验和 16SrRNA 寡核苷酸编目的相似性比较 rRNA 顺反子的实验数据可得到属以上细菌分类单元的较一致的系统发育概念,并导致了古细菌的建立。
4 、 16S rRNA(16S rDNA) 寡核苷酸的序列分析
首先, 16S rRNA 普遍存在于原核生物(真核生物中其同源分子是 18S rRNA )中。 rRNA 参与生物蛋白质的合成过程,其功能是任何生物都必不可少的,而且在生物进化的漫长历程中保持不变,可看作为生物演变的时间钟。其次,在 16S rRNA 分子中,既含有高度保守的序列区域,又有中度保守和高度变化的序列区域,因而它适用于进化距离不同的各类生物亲缘关系的研究。第三, 16S rRNA 的相对分子量大小适中,约 1 540 个核苷酸,便于序列分析。因此,它可以作为测量各类生物进化和亲缘关系的良好工具。
分离菌株 16S rRNA 基因的分离较为简单。从平板中直接挑取一环分离菌株细胞 , 加入 100μL 无菌重蒸 H 2 O 中 , 旋涡混匀后 , 沸水浴 2min, 12 000r min -1 离心 5min, 上清液中即含 16S rRNA 基因,可直接用于 PCR 扩增。分离菌株 16S rRNA 基因的 PCR 扩增和序列测定的一般步骤为: 16S rRNA 基因的 PCR 引物: 5"-AGAGT TTGAT CCTGG CTCAG-3" ; 5"-AAGGA GGTGA TCCAG CCGCA-3" 。扩增反应体积 50 m L ,反应条件为 95 ℃预变性 5min , 94 ℃变性 1min , 55 ℃退火 1min , 72 ℃延伸 2min ,共进行 29 个循环, PCR 反应在 PTC-200 型热循环仪上进行。取 5 m L 反应液在 10g L -1 的琼脂糖凝胶上进行电泳检测。 PCR 产物测序可由专门技术公司完成。
测序得到 分离菌株 16S rDNA 部分序列,此序列一般以 *.f.seq 形式保存,可以用写字板或 Editsequence 软件打开,将所得序列通过 Blast 程序与 GenBank 中核酸数据进行比对分析 ( http://www.ncbi.nlm.nih.gov/blast ) ,具体步骤如下:点击网站中 Nucleotide BLAST 下 Nucleotide-nucleotide BLAST [blastn] 选项,将测序所得序列粘贴在“ search ”网页空白处,或输入测序结果所在文件夹目录,点击核酸比对选项,即“ blast ”,然后点击“ format ”,计算机自动开始搜索核苷酸数据库中序列并进行序列比较,根据同源性高低列出相近序列及其所属种或属,以及菌株相关信息,从而初步判断 16S rDNA 鉴定结果。
遗传距离矩阵与系统发育树构建,可采用 DNAStar 软件包中的 MegAlign 程序计算样本间的遗传距离。由 GenBank 中得到相关菌株的序列,与本研究分离菌株所测得序列一起输入 Clustalx1.8 程序进行 DNA 同源序列排列,并经人工仔细核查。在此基础上,序列输入 Phylip3.6 软件包,以简约法构建系统发育树。使用 Kimura 2-parameter 法,系统树各分枝的置信度经重抽样法( Bootstrap ) 500 次重复检测, DNA 序列变异中的转换和颠换赋于相同的加权值。
上一篇:微生物鉴定的技术与方法—遗传学分类法
下一篇:一株植物病原真菌拮抗细菌的分离与鉴定
分享到:
 收藏
收藏
收藏
评论排行
- ·中国发酵企业数据库(4)
- ·(4)
- ·CoQ10高产菌株选育的研究进展(2)
- ·抗生素发酵工艺所用冷却塔的性能分析及处理(1)
- ·微生物菌种选育技术.rar(1)
- ·发酵生产染菌及其防治(1)
- ·赤藓糖醇发酵工艺研究(1)
- ·重组AiiA 蛋白可溶性表达及发酵条件优化(1)
- ·生物反应器设计软件_发酵罐绿色版(1)
- ·酵母粉、酵母浸粉的区别(1)
- ·雷帕霉素研究进展(1)
- ·透明质酸用途和行业概况(1)
- ·黄酒制作工艺(1)
- ·水解(酸化)工艺与厌氧发酵的区别(1)
- ·糖蜜酒精废液处理过程中产生的微生物蛋...(1)
- ·紫杉醇高产菌发酵产物的分离、纯化和鉴定(1)